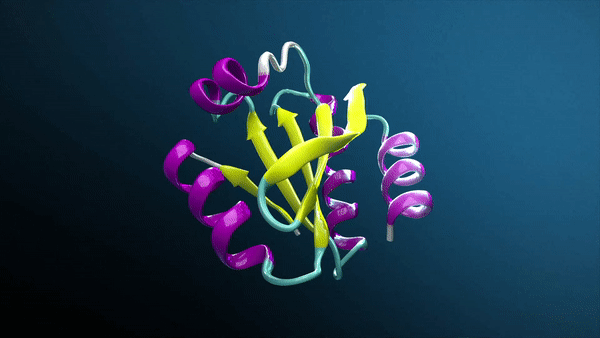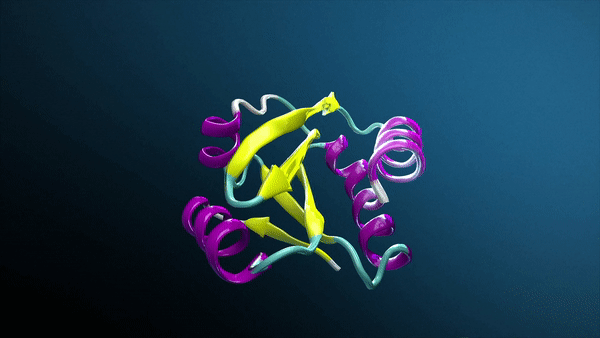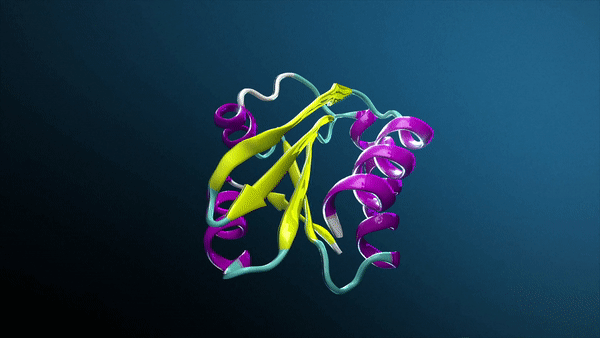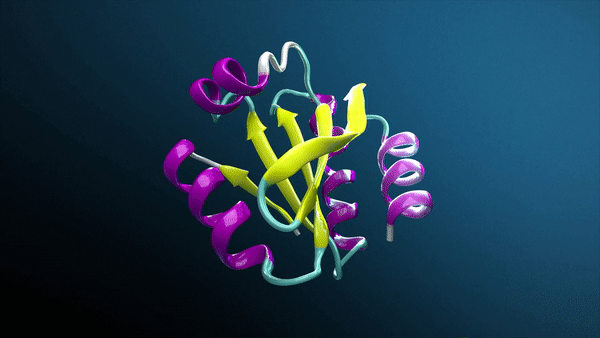
Generative AI, especially with breakthroughs like AlphaFold and RosettaFold, is transforming drug discovery and how biotech companies and research laboratories study protein structures, unlocking groundbreaking insights into protein interactions.
Proteins are dynamic entities. It has been postulated that a protein’s native state is known by its sequence of amino acids alone, as in AlphaFold2 and Its Applications in the Fields of Biology and Medicine. Yet a single protein can adopt multiple distinct conformations that can be seen depending on the environment, for example, a change in temperature. These conformations can reflect flexible regions, interactions within protein complexes, or transitions between active and inactive states.
Protai, a member of the NVIDIA Inception program for startups, aims to capture the structural changes between different protein states to determine the most precise protein structure for a specific mechanism of action (MOA), rather than settling for one conformation that might not be the most therapeutically relevant.
Protai is pushing the boundaries of drug discovery by leveraging Mass Spectrometry Proteomics and AI to develop precision medicine solutions that make a real difference for human health and society. At the core of Protai’s platform is a protein structure prediction pipeline, which integrates Nobel-winning protein structure algorithms, physics-based tools, and proprietary proteomics data.

To accelerate AI inferencing on their platform, Protai adopted NVIDIA NIM microservices for drug discovery, a set of optimized generative AI biology models. With NIM microservices, Protai achieved significantly higher throughput and lower latency for their protein structure predictions without compromising accuracy. This post dives into how Protai integrates NVIDIA NIM to power accurate and scalable protein structure predictions, transforming how they approach drug discovery.
Protein complex structure prediction background
Understanding protein complexes is a fundamental pillar of structural biology. A protein complex is a group of two or more associated polypeptide chains that interact and function together to perform specific biological activities. Multimeric proteins—assemblies of multiple interacting proteins—drive critical biological processes and are key targets in drug discovery. While the scientific community is making progress in determining the structure of every protein monomer, based on either experimental or computational techniques, the number of protein complexes is exponentially larger. This highlights the need for prediction algorithms to facilitate structural work on these complexes.
AlphaFold-Multimer fills this gap by enabling high-quality computational predictions of multimeric protein structures. This innovation, built on the foundation of AlphaFold, uses deep learning to decipher inter-protein interactions. We provide more details about the AlphaFold algorithm, parameters, outputs, and deployment in a later section.
Protai employs a multifaceted approach. One key strategy involves enhancing the sampling of structural prediction models, such as AlphaFold-Multimer, to account for structural shifts. Additionally, Protai generates unique cross-linking mass spectrometry (XL-MS) data to identify linkers that reveal specific protein structures in distinct states.
XL-MS is a powerful experimental technique that uses chemical cross-linkers to covalently bond specific amino acid residues within or between proteins, capturing spatial proximity and interaction sites. These cross-linked regions provide valuable distance constraints, enabling a more precise mapping of protein conformations and interactions. By combining these experimentally derived constraints with advanced sampling techniques and molecular dynamics simulations, researchers can generate protein structures that go beyond what’s currently available in the public domain.
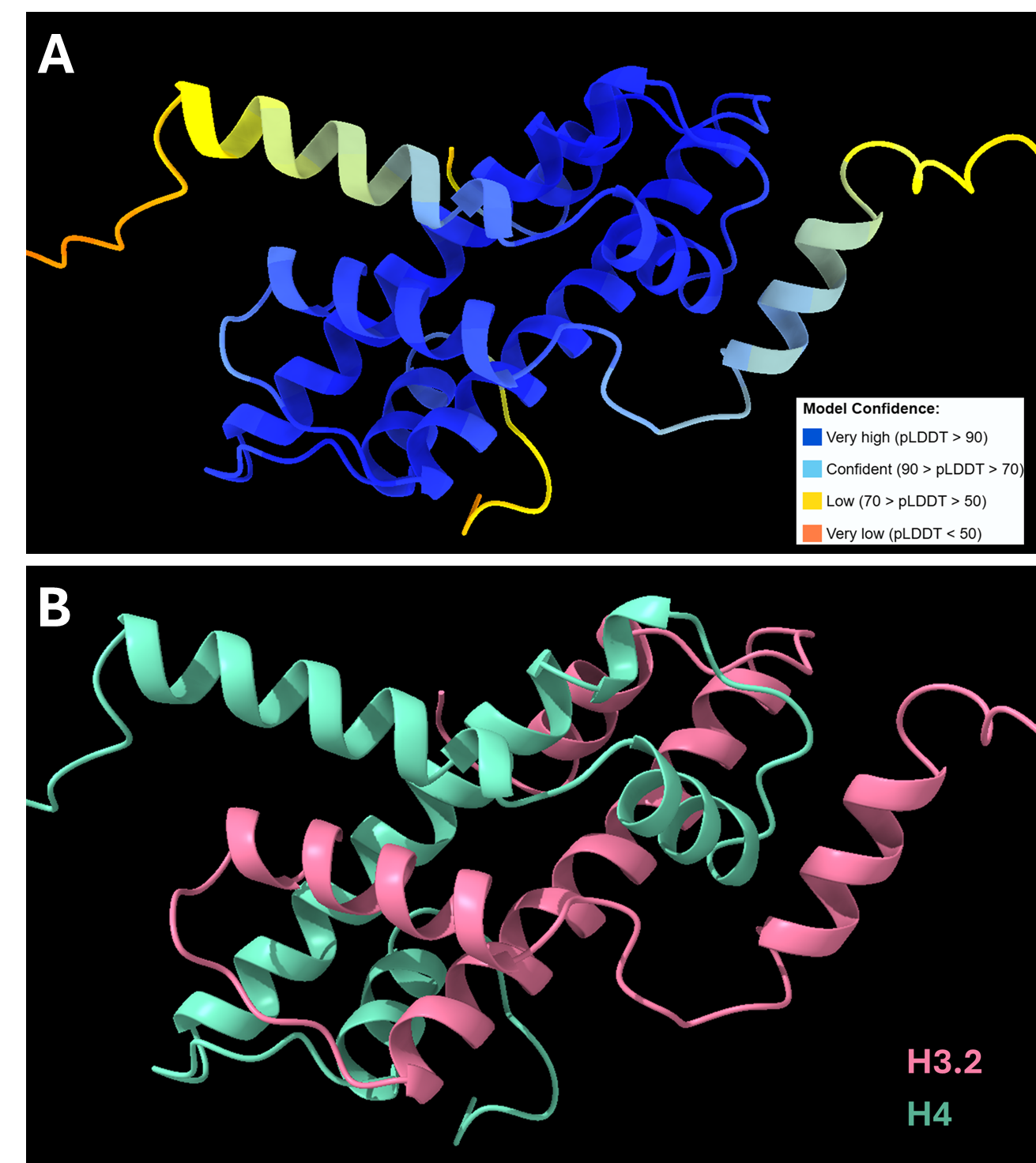
Case study: Predicting the H3-H4 protein complex
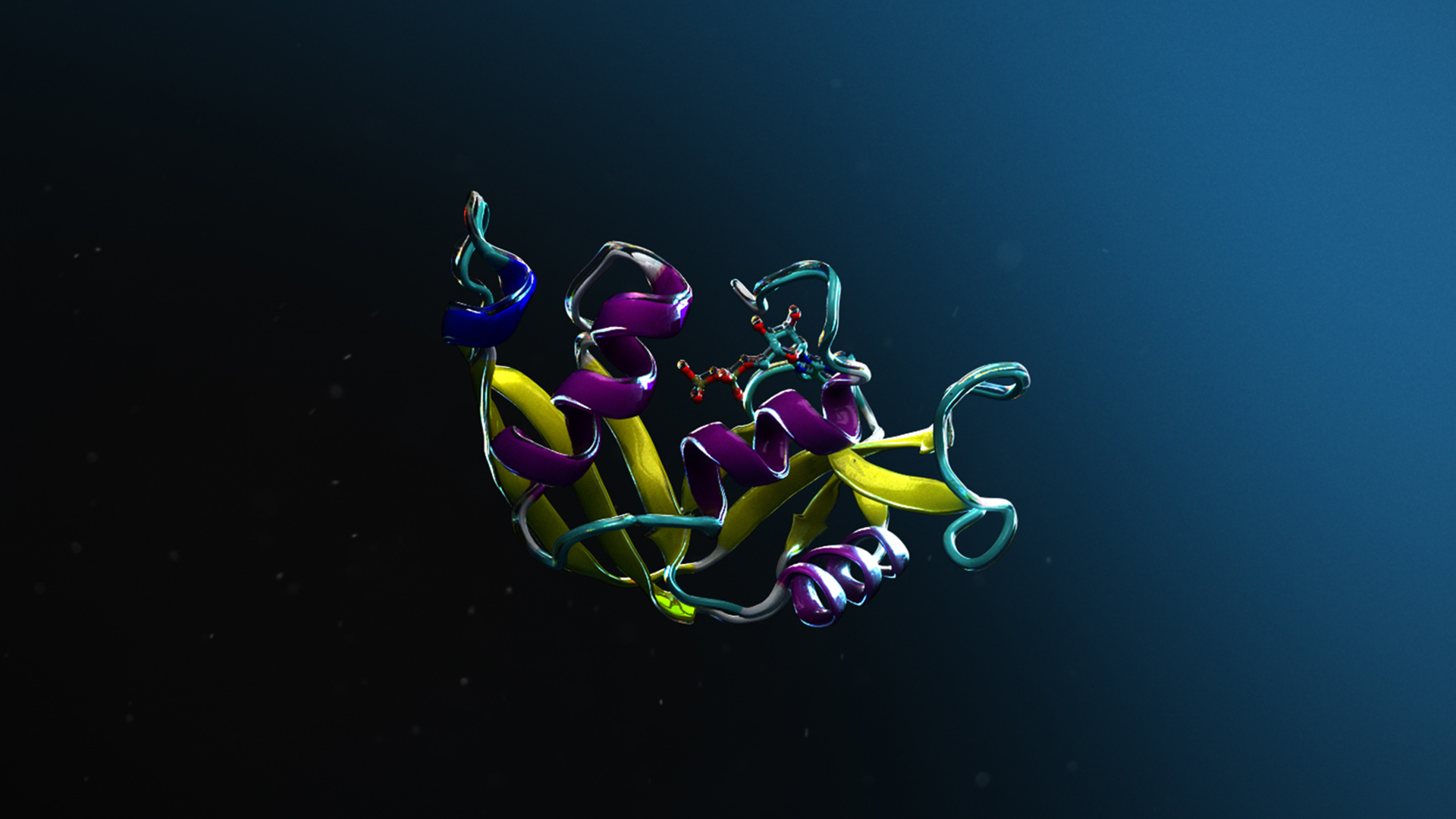
To illustrate Protai’s capabilities, this section examines the prediction of a protein complex involving histones H3 and H4. This complex plays a vital role in determining DNA accessibility to transcription factors and RNA polymerase, while also contributing to DNA stability during repair processes.
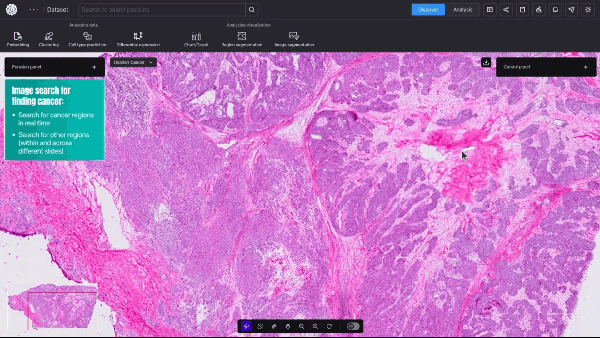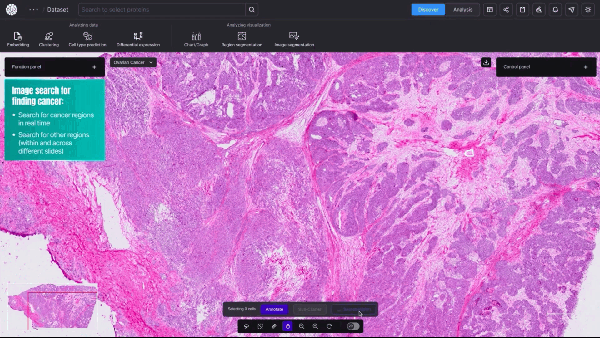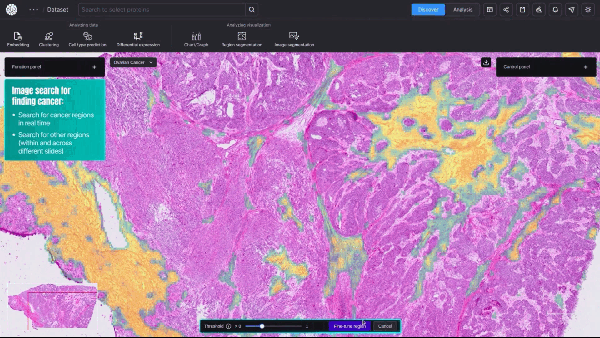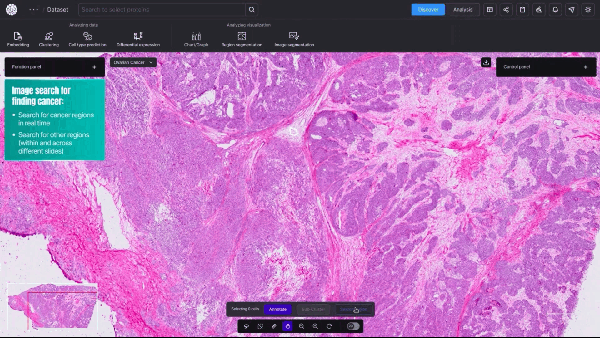
Using AlphaFold2-Multimer NIM, Protai generated a structural prediction of the H3-H4 complex. The resulting structure is color-coded by confidence, providing a visual representation of prediction accuracy. For proteins included in the AlphaFold training set, predictions tend to be highly confident, but flexibility and unique features can vary depending on the specific conformation.
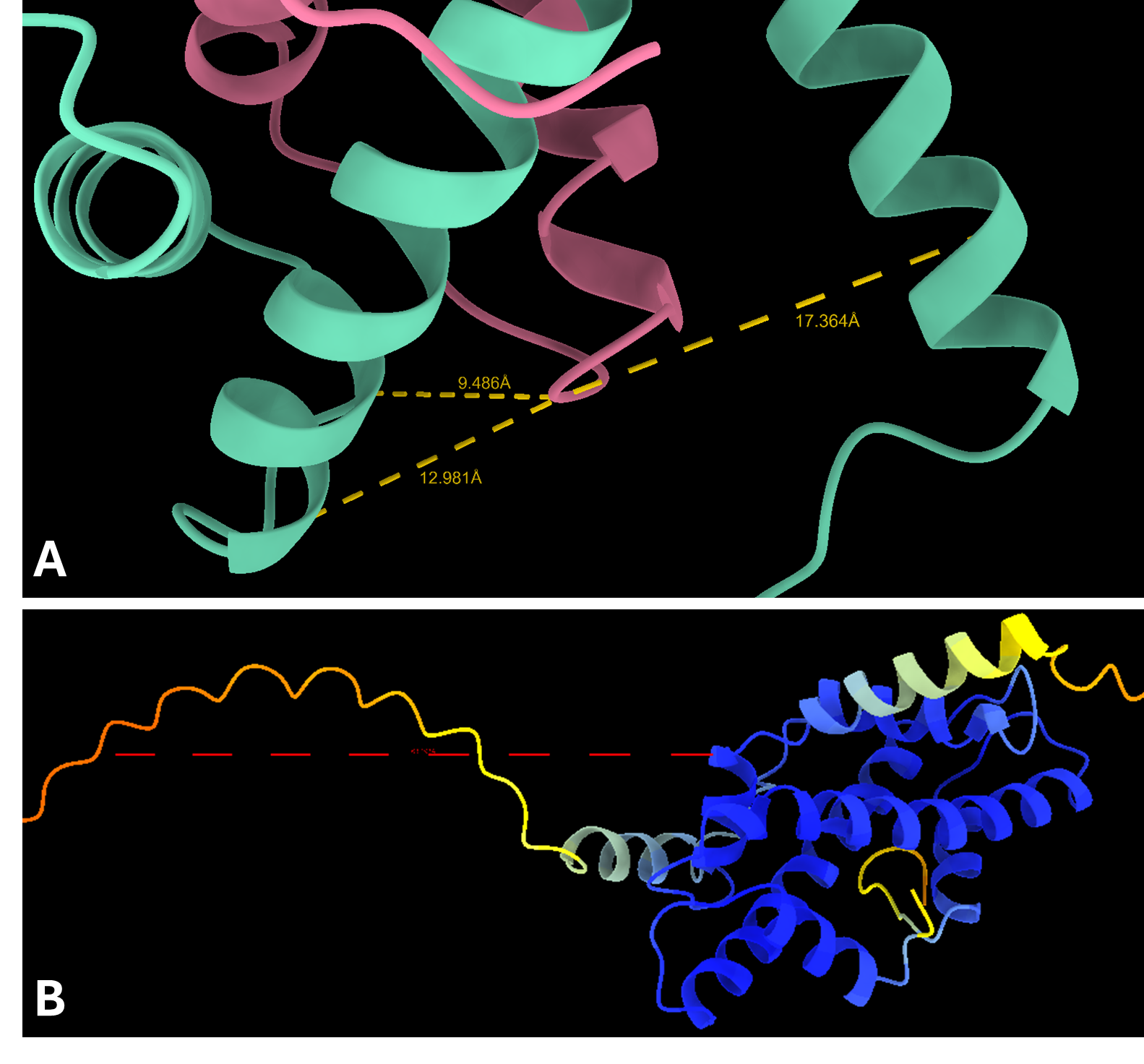
To refine these predictions, Protai leveraged XL-MS data, identifying three inter-protein linkers between H3 and H4. These linkers serve as experimental anchors, enabling Protai to validate the predicted structure or uncover new potential protein states. In this case, three linkers were held consistently across the top five ranked predictions. An additional linker was identified between regions of high and low confidence residues in the two proteins, highlighting an opportunity to further refine and improve the prediction.
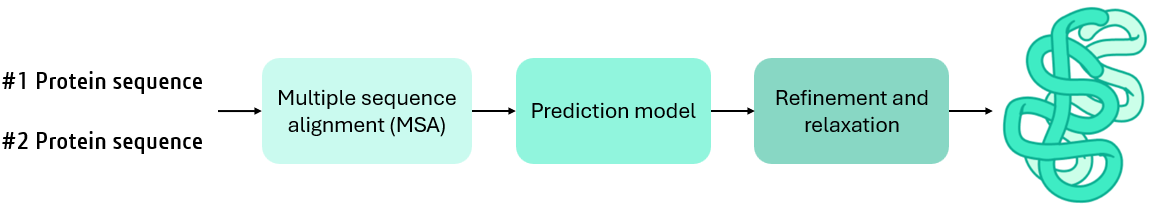
AlphaFold-Multimer deployment with NVIDIA NIM
To support scalable and optimized deployment of AlphaFold-Multimer and other LLM-based tools, Protai leverages NVIDIA NIM. NIM provides preoptimized inference containers for seamless deployment on NVIDIA GPUs. These containers ensure state-of-the-art performance, whether running on-premises or in the cloud. AlphaFold itself cannot utilize multiple GPUs for a single prediction. However, the NIM microservice enables running multiple inference tasks in parallel, significantly reducing the overall time required for predicting multiple protein complexes. (For large complexes, the prediction could take more than 24 hours.)
The workflow consists of the following key stages:
- Multiple Sequence Alignment (MSA): An MSA identifies conserved regions and co-evolutionary signals between interacting proteins, providing a foundation for predictions. Traditional MSA tools rely on CPU-based implementations, which, while effective at sequential processing, can’t match GPU parallel processing capabilities. AlphaFold2 NIM uses MMseqs2, a GPU-optimized sequence search and clustering suite achieving efficient comparisons at unparalleled speed.
- Modeling Protein Interactions: AlphaFold-Multimer uses a modified version of the AlphaFold2 transformer-based architecture, fine-tuned for interchain interactions, enhanced pair representations, cross-chain modeling, and multimer-specific loss functions to predict protein complex structures.
- Structure Refinement: The predicted structures undergo refinement to ensure accuracy and physical plausibility, incorporating stereochemical constraints and experimental benchmarks.
AlphaFold2-Multimer NIM offers multiple endpoints that enable splitting the inference process into CPU-intensive and GPU-intensive tasks, reducing computational costs:
- protein-structure/alphafold2/multimer/predict-structure-from-sequences: Full structure prediction from sequence (end-to-end)
- protein-structure/alphafold2/multimer/predict-MSA-from-sequences: MSA computation from sequence (CPU-intensive)
- protein-structure/alphafold2/multimer/predict-structure-from-MSA: Structure prediction from a precomputed MSA (GPU-intensive)
The choice of hardware depends on the size of the protein complex:
- Short sequences: A GPU with 32 GB is sufficient.
- Larger complexes (>3,000 residues): NVIDIA H100 or A100 GPUs are required for optimal performance.
For the use case above, Protai self-deployed the AlphaFold2-Multimer NIM on an NVIDIA L4 GPU, as the two proteins were small.
Parameters
AlphaFold2-Multimer NIM features the following parameters:
- sequences: Defines the target protein chains for which the multimeric structure prediction will be performed.
- algorithm: Specifies the algorithm used to search for homologous sequences. jackhmmer is a widely used algorithm for MSA generation, which identifies evolutionarily related sequences that help the model learn inter-protein interactions.
- e_value: A threshold for identifying homologous sequences in the databases. Lower values indicate stricter criteria for matches.
- iterations: Defines the number of iterations the MSA algorithm performs (limits runtime).
- databases: Specifies the databases queried for evolutionary information required for accurate MSA.
- relax_prediction: Indicates whether the predicted structure should be refined. Ensures the physical plausibility of the final structure by optimizing bond lengths, angles, and stereochemical constraints.
Output
The AlphaFold2-Multimer NIM output in the Protein Data Bank (PDB) format contains atomic-level structural information about predicted protein multimers. In the output file, each atom of the protein is described using a structured format that adheres to the PDB format specification.
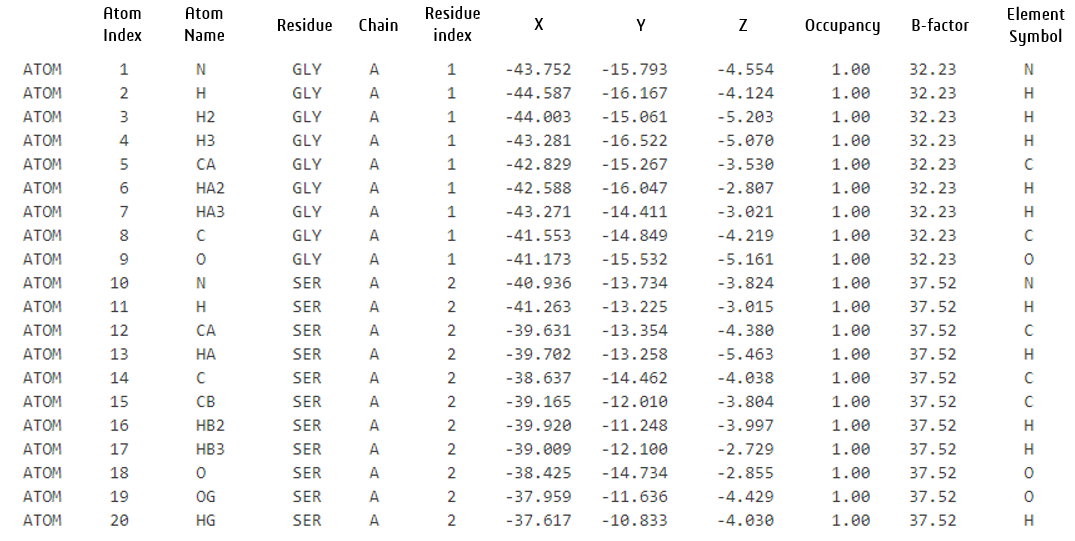
The given PDB lines using the AlphaFold2-Multimer NIM are explained below:
- Chain identifier: Used to identify the protein chain in the output, for example a protein complex of two proteins will have at least two chains.
- Coordinates (X, Y, Z): The 3D spatial coordinates of the atom in angstroms (Å).
- Occupancy: The probability that this atom is observed in this position. It ranges from 0.00 (completely absent) to 1.00 (always present). In experimental data, if an atom appears in multiple conformations in a crystal structure, its occupancy might be less than 1.00 (e.g. side chains with multiple rotamers (alternate conformations) may have occupancies summing to 1.00 across all conformations). In AlphaFold predictions, occupancy is set to 1 for all atoms because it provides a single most likely structure for the protein.
- B-factor: A measure of atomic displacement or flexibility in the structure. Higher values indicate more movement. In experimental data it can represent thermal motion or disorder. In AlphaFold outputs it is replaced by a confidence score derived from the Predicted Local Distance Difference Test (pLDDT). pLDDT is a per-residue confidence score provided by AlphaFold that ranges from 0 to 100.
Conclusion
Protai’s structure prediction workflow combines the AlphaFold2-Multimer NIM, together with unique XL-MS linkers identified experimentally. By leveraging the NVIDIA optimized AI infrastructure, Protai accelerated predictions and improved scalability. This enables exploration of previously inaccessible protein interactions, opening new frontiers in drug discovery and precision medicine.
The H3-H4 test case demonstrates how Protai’s integrative approach—merging structural prediction and XL-MS data—can unlock insights into protein conformations and dynamics that are critical for understanding biological function and therapeutic applications.
As generative AI continues to evolve, tools like NVIDIA NIM and NVIDIA BioNeMo Framework will play a pivotal role in unlocking the full potential of computational biology. Protai remains committed to integrating cutting-edge AI technologies, bringing us closer to a future where life-saving drugs are developed faster, more efficiently, and with greater precision than ever before.
You can experiment with the full set of NIM microservices for drug discovery, including the NVIDIA BioNeMo Blueprint for generative protein binder design and the NVIDIA BioNeMo Blueprint for generative virtual screening. And you can go further by training your own biology models with the open-source BioNeMo Framework. Finally, learn how to boost AlphaFold2 protein structure prediction with GPU-accelerated MMseqs2.